We show that RNA editing sites can be called with high confidence using RNA sequencing data from multiple samples across either individuals or species without the need for matched genomic DNA sequence. Milos1 Nature Reviews Genetics 12 87-98 February 2011 Computational methods for transcriptome annotation and quantification using RNA-seq Manuel Garber Manfred G Grabherr Mitchell Guttman Cole Trapnell Nature Methods 8 469477 2011.

Pin On Cd Genomics
Cancer RNA-Seq to Detect Transcriptome Changes.

. We chose the length of the splice junction regions to be slightly. We use the Clontech Ultra Low Input RNA SMARTer mRNA amplification kit for the initial RNA amplification which provides a fast and simple method for preparing amplified cDNA from total RNA for RNA-Seq applications. NUSeq generates single-end 50 or 75 bp reads for small RNA-seq.
A Schematic of the IKZF1 canonical transcript IK1 and augmented deletion transcripts del4-7 del4-8 del2-7 and del2-8. However the diversity of modifications found in RNA poses a. We identified many previously unidentified editing sites in both humans and Drosophila.
Spearman correlation analysis revealed that DV200 associated best with most if not all RNA sequencing quality parameters compared to DV100 DV150 and RIN across all FFPE samples analyzed. With the ability to sequence whole genomes identifying novel changes between individuals to exploring what RNA sequences are being expressed or to examine DNA modifications and protein-DNA interactions occurring that can help researchers better. MRNA sequencing mRNA-Seq has rapidly become the method of choice for analyzing the transcriptomes of disease states of biological processes and across a wide range of study designs.
The suggested sequencing depth is 4-5 million reads per sample. RNA-seq libraries were prepared and sequenced on the Illumina HiSeq instrument. Cancer transcriptome sequencing captures both coding.
RNA sequencing RNA-Seq is revolutionizing the study of the transcriptome. Low-input or ultra-low-input RNA-seq. Identifying IKZF1 deletions using RNA-seq data.
Our results provide a probabilistic framework for. The Center for Medical Genomics considers a variety of factors to decide which RNA protocol to use. Sequencing depth may be reduced to some extent based on the amount of starting material.
Notably DV200 correlated far better than RIN for global gene detection ρ 082 and ρ 015 respectively. Why is Next Generation Sequencing so powerful to explore and answer both clinical and research questions. Advances challenges and opportunities Fatih Ozsolak1 Patrice M.
The major challenge in identifying RNA editing sites using RNA-seq data is the. Read length remains the same as standard mRNA- or total RNA-seq. High-throughput RNA sequencing RNA-seq has enabled transcriptome-wide identification of A-to-I editing sites.
QC Metric Guidelines mRNA total RNA RNA Types Coding Coding non-coding RIN 8 low RIN 3 bias 8 Single-end vs Paired-end Paired-end Paired-end Recommended Sequencing Depth 10-20M PE reads 25-60M PE reads FastQC Q30 70 Q30 70 Percent Aligned to Reference 70 65 Million Reads Aligned Reference 7M PE reads or 14M reads 165M PE reads. Total RNA-seq Normally total RNA-Seq also known as whole-transcriptome sequencing refers to the sequencing of RNA that has been depleted of ribosomal RNA rRNA which represents the majority of RNA molecules both coding and noncoding. In addition to being a highly sensitive and accurate means of quantifying gene expression mRNA-Seq can identify both known and novel transcript.
To detect alternative splicing using RNA-seq data MISO and other methods use sequence reads aligned to splice-junction sequences that are either precomputed from known or predicted exon-intron boundaries or discovered de novo by spliced alignment to the genome Online MethodsIn the most common type of alternative splicing in. The obtained dscDNA is then converted into a sequencing library using Kapa Hyper Prep Kit. RNA-seq is an approach to estimate transcript abundance by sequencing the transcriptome of a cell type or tissue.
We offer best-in-class tools for rapid and accurate transcriptome analysis using RNA-seq and are continuously refining and building upon our core SMART S witching M echanism a t the 5 end of R NA T. Reads were filtered and aligned to the hg19 human reference genome using TopHat. The exons encoding the N-terminal DNA-binding and C-terminal dimerization domains are indicated by arrows.
RNA sequencing RNA-seq can not only be used to identify the expression of common or rare transcripts but also in the identification of other abnormal events such as alternative splicing novel transcripts and fusion genes. A highly sensitive and accurate tool for measuring expression across the transcriptome it is providing researchers with visibility into previously undetected changes occurring in disease states in response to therapeutics under different environmental conditions and across a broad range of other. Sequencing the coding regions or the whole cancer transcriptome can provide valuable information about gene expression changes in tumors.
Introduction to mRNA Sequencing. Cancer RNA-Seq enables detection of strand-specific information an important component of gene regulation. Good science leaves no possibility uninvestigatedno matter how small the sample size or how varied the sample composition.
Much progress has been made in using nanopores for DNA sequencing 313233 and identifying several DNA modifications 343536373839. It is primarily used to identify genes that are either upregulated or downregulated in relation to a control. Second step normalization was carried out to compare gene expression values FPKM across samples using a normalization factor derived from 18 reference genes.
Miso implicated the rnA splicing factor hnrnP h in the regulation of alternative cleavage and polyadenylation a role that was supported by uV cross-linkingimmunoprecipitation sequencing cliP-seq analysis in human cells. We adopted our previously published pipeline to accurately map RNA-seq reads onto the genome. 4 This can give researchers vital information about the.
Each coding exon is colored and noncoding exons are gray. In brief we used the Burrows-Wheeler Algorithm BWA 23 to align RNA-seq reads to a combination of the reference genome and exonic sequences surrounding known splice junctions from available gene models. Quantifying alternative splicing with MISO.
Some of the most popular techniques that use RNA-seq are transcriptional profiling single nucleotide polymorphism SNP identification 3 RNA editing and differential gene expression analysis.

Experimental Workflow For Rna Seq Using The Ion Torrent See Details In Download Scientific Diagram

Sequencing Instruments Genomescan
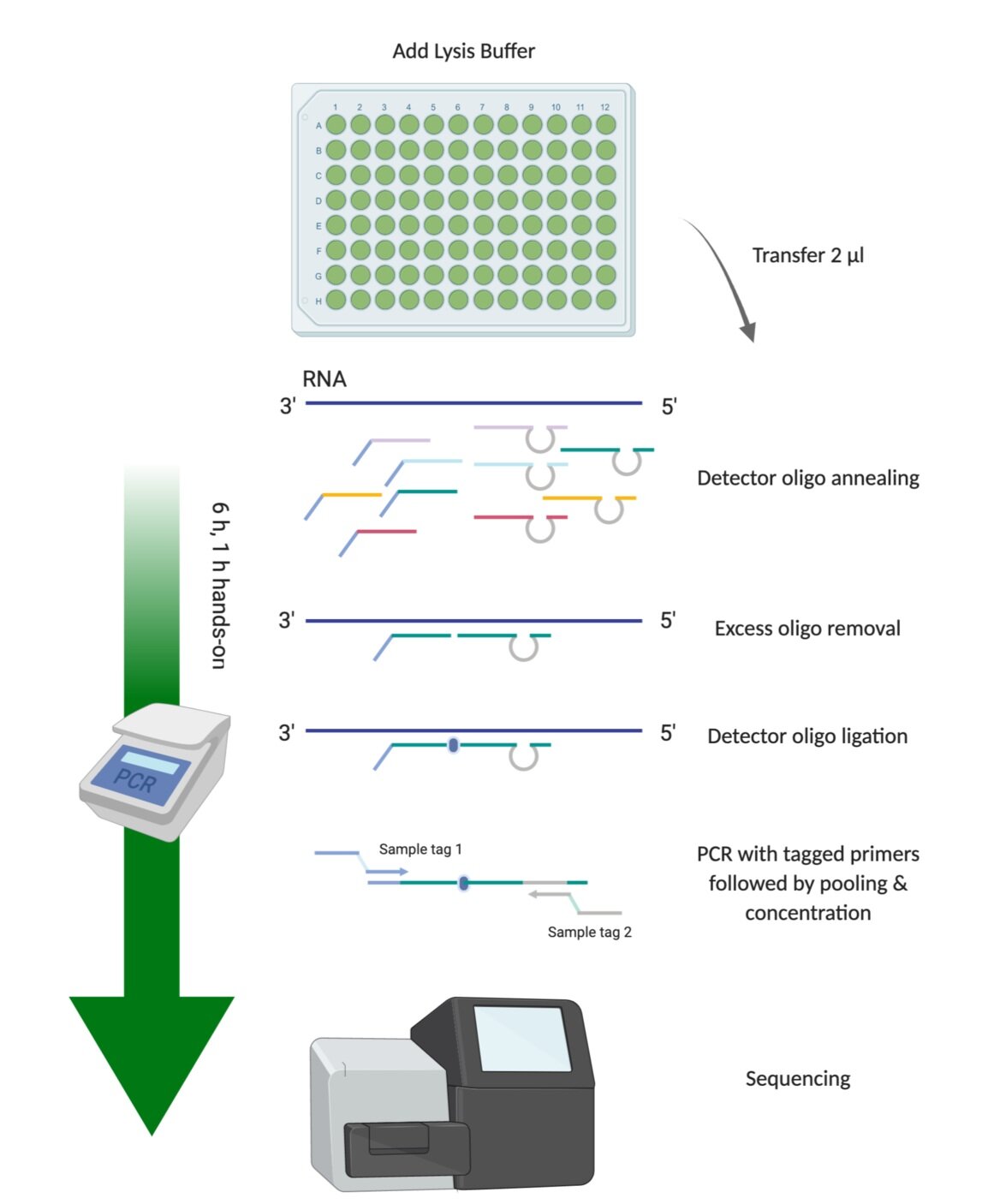
Tempo Seq Workflow Biospyder
Sequencing Instruments Genomescan
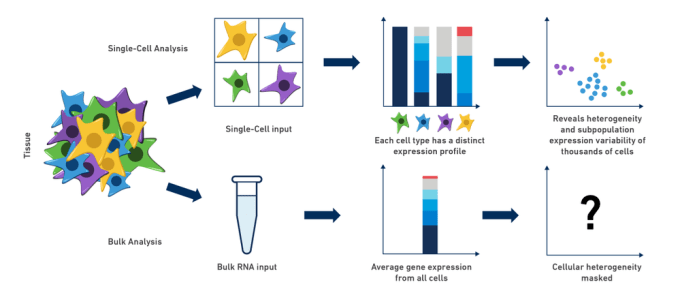
Single Cell Rna Seq An Introductory Overview And Tools For Getting Started 10x Genomics

Sequencing Instruments Genomescan

Experimental Workflow For Rna Seq Using The Ion Torrent See Details In Download Scientific Diagram

Bioinformatics Services Next Generation Sequencing Service Data Analysis

Single Cell Rna Sequencing Scipio





0 Comments